
Cusabio N-terminal 10xHis-HA-tagged Recombinant
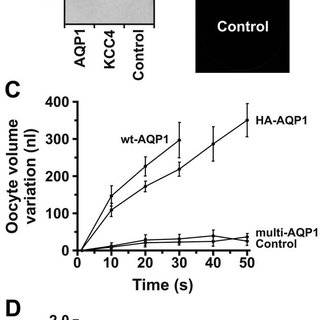
What is an HA tag? An HA tag is derived from the human influenza hemagglutinin (HA) molecule corresponding to amino acids 98-106. It has been widely used as a general epitope tag in expression vectors. The HA tag does not appear to interfere with the bioactivity or biodistribution of the recombinant HA-tagged protein. What are … Read more


![[Novel vector preS1-tp fusion protein effectively inhibits hepatitis B virus replication and cccDNA synthesis by mediating hepatitis B virus targeting sequence small interfering RNA]](http://maizetedb.org/wp-content/uploads/2021/03/IMG-20201217-WA0048.jpg)


