
Genome-scale DNA methylation maps of pluripotent and differentiated cells.
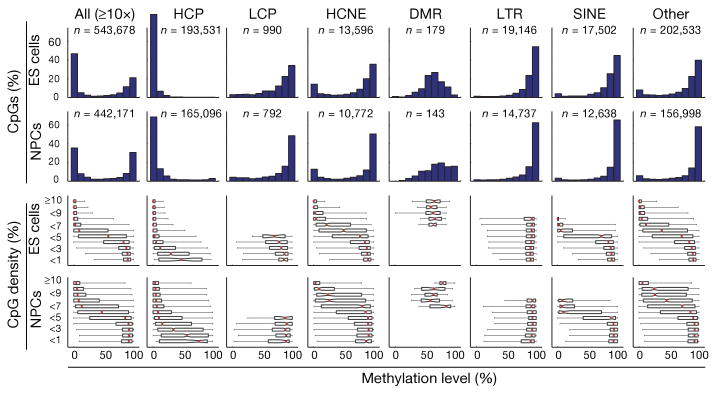
DNA methylation is important for regular growth and has been implicated in lots of pathologies together with most cancers. Our information concerning the genome-wide distribution of DNA methylation, the way it modifications throughout mobile differentiation and the way it pertains to histone methylation and different chromatin modifications in mammals stays restricted. Here we report the … Read more