
DNA methylation is important for regular growth and has been implicated in lots of pathologies together with most cancers.
Our information concerning the genome-wide distribution of DNA methylation, the way it modifications throughout mobile differentiation and the way it pertains to histone methylation and different chromatin modifications in mammals stays restricted.
Here we report the era and evaluation of genome-scale DNA methylation profiles at nucleotide decision in mammalian cells.
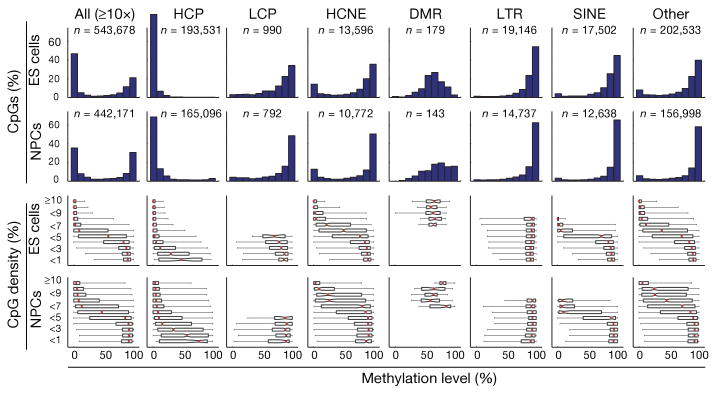
Using high-throughput decreased illustration bisulphite sequencing and single-molecule-based sequencing, we generated DNA methylation maps overlaying most CpG islands, and a consultant sampling of conserved non-coding components, transposons and different genomic options, for mouse embryonic stem cells, embryonic-stem-cell-derived and main neural cells, and eight different main tissues.
Several key findings emerge from the information. First, DNA methylation patterns are higher correlated with histone methylation patterns than with the underlying genome sequence context.
Second, methylation of CpGs are dynamic epigenetic marks that bear intensive modifications throughout mobile differentiation, significantly in regulatory areas outdoors of core promoters. Third, evaluation of embryonic-stem-cell-derived and main cells reveals that ‘weak’ CpG islands related to a particular set of developmentally regulated genes bear aberrant hypermethylation throughout prolonged proliferation in vitro, in a sample reminiscent of that reported in some main tumours.
More usually, the outcomes set up decreased illustration bisulphite sequencing as a strong expertise for epigenetic profiling of cell populations related to developmental biology, most cancers and regenerative medication.
Distribution, silencing potential and evolutionary impression of promoter DNA methylation within the human genome.
To acquire perception into the operate of DNA methylation at cis-regulatory areas and its impression on gene expression, we measured methylation, RNA polymerase occupancy and histone modifications at 16,000 promoters in main human somatic and germline cells. We discover CpG-poor promoters hypermethylated in somatic cells, which doesn’t preclude their exercise.
This methylation is current in male gametes and leads to evolutionary loss of CpG dinucleotides, as measured by divergence between people and primates. In distinction, robust CpG island promoters are largely unmethylated, even when inactive. Weak CpG island promoters are distinct, as they’re preferential targets for de novo methylation in somatic cells. Notably, most germline-specific genes are methylated in somatic cells, suggesting extra useful choice.
These outcomes present that promoter sequence and gene operate are main predictors of promoter methylation states. Moreover, we observe that inactive unmethylated CpG island promoters present elevated ranges of dimethylation of Lys4 of histone H3, suggesting that this chromatin mark might defend DNA from methylation.